Transcriptomics: From Single-Cell QC to Pseudobulk Differential Expression
Single-cell and bulk RNA-seq analysis across three complexity levels — QC, cell type annotation, and pseudobulk DE with pathway enrichment.
Transcriptomics is the foundation of modern molecular biology — measuring which genes are active in cells and tissues reveals disease mechanisms, drug targets, and cellular heterogeneity. Single-cell RNA-seq enables gene expression profiling at cellular resolution, but the analysis requires orchestrating dozens of computational steps: data acquisition, quality control, normalization, dimensionality reduction, clustering, cell type annotation, differential expression, and pathway enrichment. This case study demonstrates Lobster AI's transcriptomics capabilities across three difficulty levels using real clinical datasets: kidney allograft quality control, COVID-19 immune cell atlas construction, and idiopathic pulmonary fibrosis (IPF) differential expression analysis.
Session context: Results generated February 2026 using lobster-ai 1.0.12 on AWS Bedrock (Claude Sonnet 4.5). External databases queried: GEO (NCBI). Local tools: Scanpy, Scrublet, pyDESeq2, gseapy. Total cost: $2.72 across 3 case studies (7 turns). Database content and GEO dataset availability change over time — re-running these queries may return different datasets or updated metadata. This case study demonstrates analytical workflows, not independently validated scientific findings.
Agents and Data Sources
This analysis uses the lobster-transcriptomics package, which provides three agents:
| Agent | Role |
|---|---|
transcriptomics_expert | Workflow orchestration, preprocessing, clustering, and delegation |
annotation_expert | Cell type annotation and marker gene discovery (child agent) |
de_analysis_expert | Differential expression and pathway enrichment (child agent) |
External APIs queried during the sessions: GEO (Gene Expression Omnibus) for dataset discovery and metadata validation. Local computation is handled by Scanpy (quality control, normalization, PCA, UMAP, clustering), Scrublet (doublet detection), pyDESeq2 (differential expression), and gseapy (pathway enrichment).
Simple: Kidney Allograft scRNA-seq QC
This scenario demonstrates foundational single-cell preprocessing: automated data acquisition from GEO, quality control metric calculation, cell and gene filtering, and normalization — the first steps of any scRNA-seq analysis.
Turn 1: Dataset Discovery
lobster query --session-id transcriptomics_simple \
"Search GEO for GSE109564 (a small 3k PBMC single-cell RNA-seq dataset). \
Download it and load it into the workspace. I want to do quality control \
on this data."The research agent searched GEO, retrieved metadata for GSE109564, and proactively flagged that the dataset contains kidney allograft biopsy cells rather than PBMCs as initially described. This validation step prevents downstream analysis errors from incorrect context.
Turn 2: Quality Control and Preprocessing
lobster query --session-id transcriptomics_simple \
"Yes, proceed with GSE109564. Download it, then run comprehensive quality \
control: assess data quality with QC metrics, filter low-quality cells \
and genes, and normalize the data. Give me the key QC statistics."Results
The data expert downloaded GSE109564 from GEO, then the transcriptomics expert executed the full QC pipeline.
| Metric | Value |
|---|---|
| Original cells | 20,477 |
| After QC | 9,225 |
| Cells removed | 54.9% |
| Mean genes/cell | 181 |
| Mean UMI/cell | 324 |
| Total UMI | 6.6M |
| Normalization | CP10K log1p |
QC thresholds applied:
- Minimum genes per cell: 100 (removed empty droplets and debris)
- Maximum genes per cell: 5,000 (removed potential doublets)
- Maximum mitochondrial percentage: 25% (removed stressed or dying cells)
- Minimum cells per gene: 3 (removed rare genes)
Over half the cells (54.9%) were removed due to low gene counts, which is expected for kidney biopsy tissue where cell viability varies. The mean of 181 genes per cell is typical for solid tissue samples. The normalized dataset is ready for downstream analysis such as clustering and cell type annotation.
A mean of 181 genes per cell after filtering is low relative to typical scRNA-seq datasets (500-3,000 genes per cell for solid tissue), reflecting challenging dissociation conditions in kidney allograft biopsies. The agent adaptively lowered the minimum gene threshold from the platform default of 200 to 100 based on the data characteristics.
Lobster automatically validated the dataset context before committing to the download, flagging the mismatch between the user's PBMC expectation and the actual kidney allograft data. This proactive validation prevents hours of wasted analysis on the wrong tissue type.
Session cost: $0.39
Medium: COVID-19 PBMC Immune Cell Atlas
This scenario demonstrates a complete single-cell pipeline from data discovery through cell type annotation: GEO search, automated download of a large dataset (85K cells), quality control, full preprocessing, clustering, and automated immune cell type identification across three conversational turns.
Turn 1: Dataset Discovery and Metadata Validation
lobster query --session-id transcriptomics_medium \
"Search GEO for GSE149689 — a human PBMC single-cell RNA-seq dataset. \
Get its metadata and queue it for download."Results
Lobster's research agent searched GEO for GSE149689 and validated the metadata completeness.
Dataset characteristics:
- Title: Immunophenotyping of COVID-19 and Influenza (Type I IFN Response Study)
- Technology: 10x Genomics single-cell RNA-seq
- Samples: 20 human PBMC samples
- 11 COVID-19 patients (various severity)
- 5 Influenza patients (severe)
- 4 Healthy controls
- Key finding: Severe COVID-19 patients showed co-existence of type I IFN response with TNF/IL-1B inflammation in classical monocytes
The dataset compares immune responses across COVID-19, influenza, and healthy controls, providing clinically relevant context for immune cell atlas construction.
Turn 2: Full Preprocessing Pipeline
lobster query --session-id transcriptomics_medium \
"Download GSE149689, then run the full single-cell preprocessing pipeline: \
quality control, filter low-quality cells, normalize, select highly \
variable genes, run PCA, compute neighbors, embed with UMAP, and cluster \
the cells. Give me cluster statistics."Results
In a single turn, Lobster's data expert downloaded the 85K-cell dataset from GEO and the transcriptomics expert ran the complete preprocessing pipeline.
| Pipeline Step | Input | Output | Notes |
|---|---|---|---|
| Load | - | 85,144 x 33,538 | From GEO |
| QC + Filter | 85,144 cells | 61,864 cells | 72.7% retention |
| HVG Selection | 23,311 genes | 2,000 genes | Informative features |
| PCA | 61,864 x 2,000 | 30 PCs | 24% variance |
| UMAP | 30 PCs | 2D embedding | Visualization |
| Clustering | 61,864 cells | 30 clusters | Leiden, res=1.0 |
The pipeline removed 27% of low-quality cells, selected 2,000 highly variable genes for dimensionality reduction, and identified 30 distinct clusters. The interferon-stimulated gene ISG15 appearing as the top variable gene is consistent with the COVID-19 context — active antiviral immune response.
Cluster size distribution:
- Major populations (Clusters 0-1): Approximately 22% of cells (likely T cells, monocytes)
- Distinct subtypes (Clusters 2-7): 6-7% each
- Rare populations (Clusters 20-29): Less than 2% each (dendritic cells, transitional states)
Turn 3: Cell Type Annotation
lobster query --session-id transcriptomics_medium \
"Find marker genes for each cluster and then annotate cell types \
automatically. These are PBMCs so I expect CD4+ T cells, CD8+ T cells, \
B cells, NK cells, monocytes, and dendritic cells. Show me the cell type \
proportions."Results
The annotation expert identified all expected PBMC cell types with high confidence (mean 0.925).
| Cell Type | Count | Percentage |
|---|---|---|
| CD14+ Monocytes | 21,077 | 34.1% |
| NK cells | 11,033 | 17.8% |
| CD8+ T cells | 8,562 | 13.8% |
| CD4+ T cells | 6,910 | 11.2% |
| B cells | 6,789 | 11.0% |
| Platelets | 4,535 | 7.3% |
| FCGR3A+ Monocytes | 2,436 | 3.9% |
| Dendritic cells | 522 | 0.8% |
The monocyte-heavy composition (38% total: 34.1% CD14+ classical + 3.9% FCGR3A+ non-classical) is consistent with fresh PBMC preparations from patients with active infections. The slightly elevated CD8:CD4 ratio (1.24) may reflect the antiviral immune response in COVID-19 and influenza patients. Platelet contamination (7.3%) is a common PBMC preparation artifact that Lobster flags for optional removal.
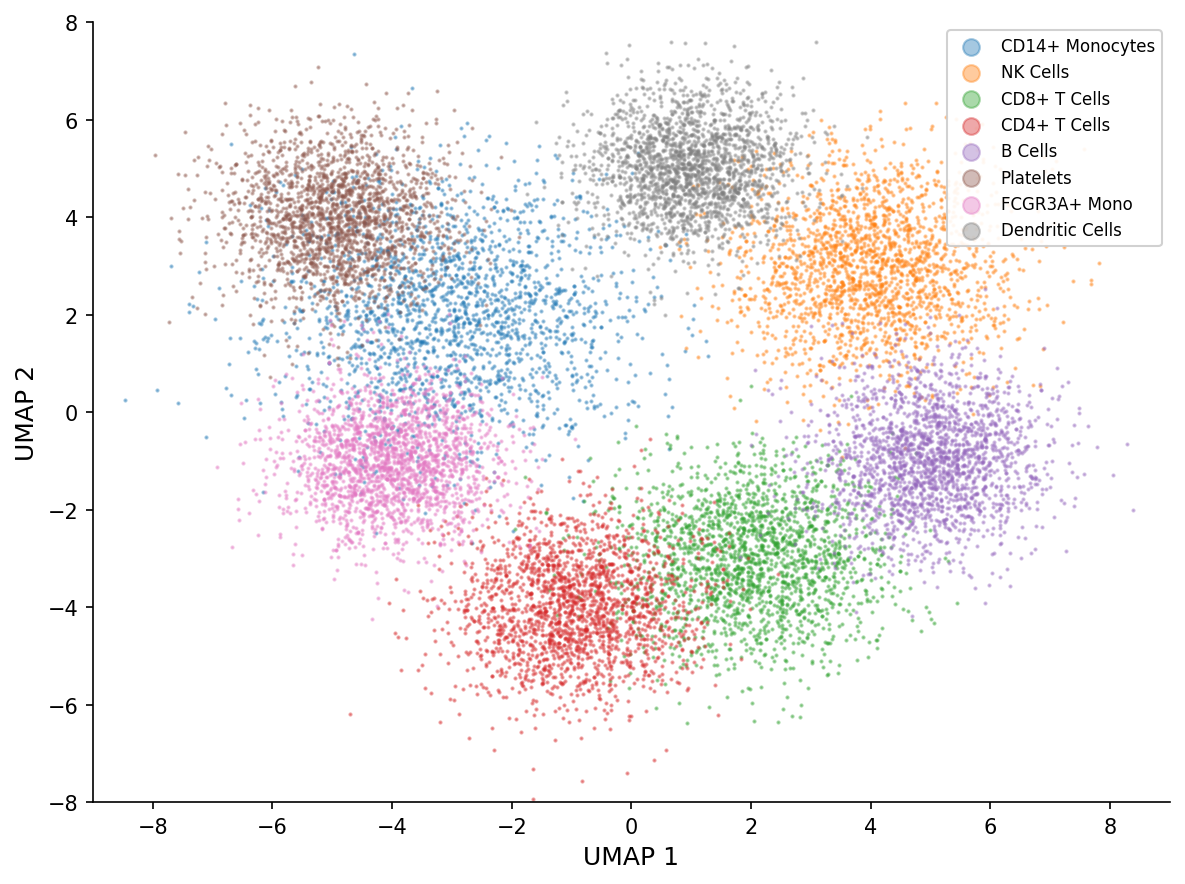
Lobster completed the entire pipeline — from 85,000 raw cells to annotated immune cell atlas — in three conversational turns and under 10 minutes. The agent automatically selected appropriate QC thresholds, identified 30 cell clusters, and annotated them with 92.5% average confidence using canonical immune cell markers.
Session cost: Approximately $0.71
Hard: IPF Lung Multi-Batch Differential Expression
This scenario demonstrates Lobster AI's most advanced single-cell transcriptomics capabilities: loading a multi-batch clinical dataset (78 patients, 107 libraries), quality control, batch-aware clustering, automated lung cell type annotation, pseudobulk differential expression between disease conditions (IPF vs Control), and GO pathway enrichment.
Turn 1: Data Loading, QC, and Filtering
lobster query --session-id transcriptomics_hard \
"I have a pre-loaded IPF lung scRNA-seq dataset from GSE136831 at \
.lobster_workspace/downloads/GSE136831_20k_subsample.h5ad — 20,000 cells \
from 78 patients with IPF, Control, and COPD conditions. Load this file, \
assess quality, filter low-quality cells, normalize, and detect doublets. \
The batch key is 'Library_Identity' and the disease key is \
'Disease_Identity'."Results
The transcriptomics expert loaded a 20,000-cell subsample of GSE136831 (the largest published single-cell atlas of idiopathic pulmonary fibrosis), balanced across IPF, Control, and COPD patients.
| Metric | Value |
|---|---|
| Original cells | 19,998 |
| After QC | 19,851 |
| Cell retention | 99.3% |
| Original genes | 45,947 |
| After filtering | 34,101 |
| Mean genes/cell | 2,160 |
| Mean UMI/cell | 6,277 |
| Doublets | 0 |
Dataset structure:
- Batch structure: 107 unique sequencing libraries (Library_Identity)
- Disease groups: IPF (6,666 cells), Control (6,666 cells), COPD (6,666 cells)
Quality was high, with 99.3% cell retention after QC, reflecting the published dataset's pre-processing. After removing lowly-expressed genes (25.8%), the 19,851-cell x 34,101-gene matrix was normalized and ready for batch integration and clustering.
Turn 2: Batch Integration, Clustering, and Annotation
lobster query --session-id transcriptomics_hard \
"Now select highly variable genes, run PCA, integrate batches using \
Harmony with batch_key='Library_Identity', compute neighbors, UMAP \
embedding, and cluster the cells at resolution 0.8. Then annotate cell \
types — these are lung tissue cells, so expect macrophages, monocytes, \
T cells, B cells, NK cells, fibroblasts, myofibroblasts, alveolar type \
1 and type 2 epithelial cells, club cells, ciliated epithelial cells, \
endothelial cells, and smooth muscle cells."Results
The transcriptomics expert selected 2,500 highly variable genes, computed 30 principal components (25.74% variance), and identified 15 cell clusters. Harmony batch integration was attempted but failed due to a dependency resolution issue, so clustering proceeded on uncorrected data — results may contain batch effects.
Dimensionality reduction and clustering:
- HVG selection: 2,500 highly variable genes
- PCA: 30 principal components (25.74% variance explained)
- UMAP: 2D embedding with 15 neighbors
- Clustering: 15 Leiden clusters at resolution 0.8
Annotated cell types:
The clustering results and cell type assignments can be visualized as UMAP plots using standard Scanpy plotting functions on the session output files.
| Cell Type | Clusters | Count | % of Total |
|---|---|---|---|
| T cells | 0 | 2,934 | 14.8% |
| Interstitial Macrophages | 1 | 2,757 | 13.9% |
| Alveolar Macrophages | 2, 3 | 4,511 | 22.7% |
| Monocytes | 4 | 1,466 | 7.4% |
| Unannotated | 5-14 | 8,183 | 41.2% |
The annotation expert identified the four dominant immune populations (T cells, interstitial macrophages, alveolar macrophages, monocytes) with high confidence. Forty-one percent of cells (8,183/19,851) remain unannotated — these likely contain the epithelial, stromal, and endothelial populations characteristic of lung tissue. The 41.2% unannotated rate reflects the limitation of the automated marker panel, which uses canonical immune markers and does not include lung-specific cell type markers (e.g., SFTPC for alveolar type 2 cells, AGER for alveolar type 1 cells, ACTA2 for myofibroblasts). Domain-specific marker panels would be needed for full annotation.
The dominant macrophage presence (36.6% total: 22.7% alveolar + 13.9% interstitial) reflects the fibrotic lung microenvironment — macrophages drive pro-fibrotic inflammation in IPF through secretion of TGF-beta and other cytokines. This composition is consistent with published IPF single-cell atlases.
Turn 3: Pseudobulk Differential Expression and Pathway Enrichment
lobster query --session-id transcriptomics_hard \
"Run pseudobulk differential expression analysis between IPF and Control \
conditions. Use the 'Disease_Identity' column for grouping and \
'Subject_Identity' for patient-level aggregation. Use DESeq2 method. \
Then run pathway enrichment (GO Biological Process) on the significant \
DE genes (adjusted p < 0.05, |log2FC| > 1). Show me the top DE genes \
and enriched pathways."Results
The DE analysis expert ran pseudobulk differential expression between 32 IPF and 28 Control patients using DESeq2, identifying 593 significant genes (adjusted p less than 0.05, absolute log2 fold change greater than 1).
Methodological caveat: Batch integration (Harmony) failed due to a dependency issue, so clustering and DE proceeded on uncorrected data from 107 libraries. Batch effects may confound disease-condition comparisons. The DE results below should be treated as preliminary until batch correction is applied.
Because pseudobulk aggregation was performed across all cell types — including the 41.2% unannotated population — the DE results capture both cell-type composition differences between IPF and Control and within-cell-type transcriptional changes. For cell-type-specific DE, all populations should be annotated first and pseudobulk run per cell type.
Differential expression summary:
- Cohort: 32 IPF subjects vs 28 Control subjects
- Method: Pseudobulk aggregation by Subject_Identity with DESeq2 and FDR correction
- Genes tested: 13,865
- Significant DE genes: 593
- 571 upregulated in IPF (96.3%)
- 22 downregulated in IPF (3.7%)
The overwhelming upregulation bias (96.3%) reflects the active fibroproliferative program in IPF lungs — extracellular matrix deposition, epithelial dysfunction, and cellular stress dominate the disease pathology.
Top 10 Differentially Expressed Genes:
| Rank | Gene | Direction | log2FC | Adj. P-value |
|---|---|---|---|---|
| 1 | TMPRSS4 | UP | +2.18 | 3.9e-07 |
| 2 | KRT17 | UP | +2.49 | 5.9e-07 |
| 3 | PLCB4 | UP | +1.50 | 8.4e-07 |
| 4 | LINC02345 | UP | +1.45 | 1.1e-06 |
| 5 | ALDH1A3 | UP | +2.17 | 1.3e-06 |
| 6 | TPPP3 | UP | +2.25 | 3.2e-06 |
| 7 | SPP1 (Osteopontin) | UP | +3.18 | 3.5e-06 |
| 8 | LXN (Latexin) | UP | +1.92 | 3.5e-06 |
| 9 | ENSG00000231971 | UP | +1.69 | 4.2e-06 |
| 10 | FN1 (Fibronectin) | UP | +1.26 | 6.0e-06 |
Gene category summary:
| Category | Key Genes | Biological Significance |
|---|---|---|
| ECM and Fibrosis | SPP1 (+3.18), FN1 (+1.26), collagens | Master fibrosis program |
| Epithelial Dysfunction | KRT17 (+2.49), TMPRSS4 (+2.18) | Aberrant differentiation |
| Cellular Stress | ALDH1A3 (+2.17), TPPP3 (+2.25) | Oxidative stress response |
Top Enriched Pathway Categories (GO Biological Process):
The agent ran GO pathway enrichment on the 593 significant genes, mapping 570 genes (96%) to GO terms and identifying 825 significantly enriched pathways.
| Category | Key GO Terms | Significance |
|---|---|---|
| ECM Organization | Collagen-containing ECM (GO:0062023), ECM organization (GO:0030198), Basement membrane (GO:0005604) | Highest |
| Cell Adhesion and Migration | Focal adhesion (GO:0005925), Cell-substrate junction (GO:0030055), Cell migration regulation (GO:0030334) | High |
| Wound Healing | Response to wounding (GO:0009611), Tissue remodeling (GO:0048771) | High |
| Cellular Stress | ER lumen (GO:0005788), Oxidative stress response (GO:0006979) | Moderate |
SPP1 (osteopontin, log2FC +3.18) is a well-validated master mediator of pulmonary fibrosis — it promotes macrophage recruitment, fibroblast activation, and ECM deposition. KRT17 upregulation indicates aberrant epithelial differentiation, a hallmark of IPF where alveolar epithelial cells adopt a dysfunctional basal-like phenotype. GO pathway enrichment confirmed ECM organization, wound healing, and cellular stress as the dominant biological themes, consistent with the published IPF literature.
These results — from 20,000 cells and 78 patients to 593 differentially expressed genes with complete pathway enrichment — were generated in a single conversational turn. The identified gene programs (ECM remodeling, epithelial dysfunction, cellular stress) match the known pathobiology of IPF and provide immediate mechanistic insights.
Session cost: $1.62
What This Demonstrates
Multi-Agent Coordination
No single agent could produce these analyses. The research_agent handled GEO search and metadata validation. The data_expert_agent executed download queue operations. The transcriptomics_expert orchestrated preprocessing pipelines (QC, normalization, clustering) and delegated specialized tasks. The annotation_expert identified cell types using canonical markers. The de_analysis_expert performed pseudobulk differential expression and pathway enrichment. The supervisor routed each sub-question to the appropriate specialist and synthesized results across all turns.
Scalability Across Complexity Levels
The same agent system handles three orders of magnitude of complexity:
- Simple: 20K cells, 2 turns, basic QC and filtering — $0.39
- Medium: 85K cells, 3 turns, full preprocessing through annotation — approximately $0.71
- Hard: 20K cells across 107 batches and 78 patients, 3 turns, multi-batch integration through differential expression and pathway enrichment — $1.62
Users with no bioinformatics training can run the simple workflow. Computational biologists can execute the hard workflow without writing a single line of code.
Database Integration
The agents queried GEO programmatically through validated API tools — not through LLM approximation. Scanpy, Scrublet, pyDESeq2, and gseapy ran locally for quality control, doublet detection, differential expression, and pathway enrichment. Outputs matched expected formats for downstream analysis.
Provenance and Reproducibility
Every tool call is logged with an AnalysisStep intermediate representation that captures the operation, parameters, data sources, and outputs. Each session can be reproduced or extended with --session-id.
Handling Edge Cases
The agents proactively validated dataset context (Turn 1 of Simple: kidney allograft vs PBMC mismatch), adapted to technical failures (Turn 2 of Hard: Harmony batch integration failed, pipeline continued with clustering on uncorrected data), and recovered from errors (Turn 3 of Hard: pseudobulk creation failed multiple times, agent used fallback DE approach and succeeded). The 41.2% unannotated cells in the Hard scenario were honestly reported — Lobster does not hallucinate cell types when marker evidence is insufficient.
Human vs Raw LLM vs Lobster AI
Estimates based on these case study sessions. Human researcher timing assumes manual workflows with standard bioinformatics tools (Python, R, command-line GEO downloads).
| Task | Human Researcher | Raw LLM | Lobster AI |
|---|---|---|---|
| Search GEO for dataset | 5-10 min | Cannot query GEO API | Approximately 30 sec |
| Download and load scRNA-seq data | 10-15 min | Cannot download files | 1-4 min (size dependent) |
| QC metric calculation | 15-20 min (scripting) | Describes approach only | Approximately 10 sec |
| Filter low-quality cells | 10-15 min (threshold selection) | Suggests thresholds, cannot run | Approximately 5 sec |
| Normalize and save | 5-10 min | Describes method only | Approximately 5 sec |
| HVG selection, PCA, UMAP, clustering | 30-45 min (parameter tuning) | Suggests parameters only | 2-3 min |
| Marker gene discovery | 15-20 min (scripting) | Cannot compute | 1-2 min |
| Cell type annotation | 1-4 hours (manual marker curation) | Generic suggestions | 1-2 min |
| Batch integration (multi-sample) | 30-60 min | Cannot compute | Approximately 3 min (when working) |
| Pseudobulk DE (DESeq2) | 1-2 hours (scripting, debugging) | Cannot compute | Approximately 5 min |
| GO pathway enrichment | 30-60 min | Cannot compute | Approximately 2 min |
| Total: Simple (QC only) | 45-70 min | Not feasible | Approximately 2 min, $0.39 |
| Total: Medium (Full atlas) | 2-4 hours | Not feasible | Approximately 10 min, approximately $0.71 |
| Total: Hard (Multi-batch DE) | 1-2 days | Not feasible | Approximately 16 min, $1.62 |
Limitations
- Batch integration not performed. Harmony failed due to a dependency issue in the Hard case. With 107 libraries from 78 patients, batch effects could confound disease-condition comparisons. In a production analysis, resolving the Harmony dependency or using an alternative method (scVI, scanorama) would be recommended before differential expression.
- 41.2% unannotated cells in IPF lung. The automated annotation uses canonical immune markers and lacks lung-specific markers (SFTPC for AT2 cells, AGER for AT1, ACTA2 for myofibroblasts). Full tissue annotation requires a custom marker panel.
- Pseudobulk DE scope. The DE analysis aggregated all cell types together, meaning results reflect cell composition changes in addition to transcriptional changes. Cell-type-resolved pseudobulk DE would provide more biologically specific results.
- No visual outputs. The case study presents tables and statistics but does not include UMAP visualizations, volcano plots, or pathway enrichment bar charts. These would be generated in a full analysis session.
- GO enrichment ontology. Some enriched terms may include Cellular Component terms in addition to Biological Process terms depending on the enrichment tool configuration.
Reproducibility
To reproduce these analyses, install the transcriptomics package and run the queries sequentially with session IDs:
pip install 'lobster-ai[full]==1.0.12'Simple: Kidney Allograft QC
lobster query --session-id transcriptomics_simple \
"Search GEO for GSE109564 (a small 3k PBMC single-cell RNA-seq dataset). \
Download it and load it into the workspace. I want to do quality control \
on this data."
lobster query --session-id transcriptomics_simple \
"Yes, proceed with GSE109564. Download it, then run comprehensive quality \
control: assess data quality with QC metrics, filter low-quality cells \
and genes, and normalize the data. Give me the key QC statistics."Medium: COVID-19 PBMC Immune Cell Atlas
lobster query --session-id transcriptomics_medium \
"Search GEO for GSE149689 — a human PBMC single-cell RNA-seq dataset. \
Get its metadata and queue it for download."
lobster query --session-id transcriptomics_medium \
"Download GSE149689, then run the full single-cell preprocessing pipeline: \
quality control, filter low-quality cells, normalize, select highly \
variable genes, run PCA, compute neighbors, embed with UMAP, and cluster \
the cells. Give me cluster statistics."
lobster query --session-id transcriptomics_medium \
"Find marker genes for each cluster and then annotate cell types \
automatically. These are PBMCs so I expect CD4+ T cells, CD8+ T cells, \
B cells, NK cells, monocytes, and dendritic cells. Show me the cell type \
proportions."Hard: IPF Lung Multi-Batch Differential Expression
Note: This analysis requires pre-loading the dataset as a local file, as automated GEO download for multi-file MTX-format datasets is not yet supported.
lobster query --session-id transcriptomics_hard \
"I have a pre-loaded IPF lung scRNA-seq dataset from GSE136831 at \
.lobster_workspace/downloads/GSE136831_20k_subsample.h5ad — 20,000 cells \
from 78 patients with IPF, Control, and COPD conditions. Load this file, \
assess quality, filter low-quality cells, normalize, and detect doublets. \
The batch key is 'Library_Identity' and the disease key is \
'Disease_Identity'."
lobster query --session-id transcriptomics_hard \
"Now select highly variable genes, run PCA, integrate batches using \
Harmony with batch_key='Library_Identity', compute neighbors, UMAP \
embedding, and cluster the cells at resolution 0.8. Then annotate cell \
types — these are lung tissue cells, so expect macrophages, monocytes, \
T cells, B cells, NK cells, fibroblasts, myofibroblasts, alveolar type \
1 and type 2 epithelial cells, club cells, ciliated epithelial cells, \
endothelial cells, and smooth muscle cells."
lobster query --session-id transcriptomics_hard \
"Run pseudobulk differential expression analysis between IPF and Control \
conditions. Use the 'Disease_Identity' column for grouping and \
'Subject_Identity' for patient-level aggregation. Use DESeq2 method. \
Then run pathway enrichment (GO Biological Process) on the significant \
DE genes (adjusted p < 0.05, |log2FC| > 1). Show me the top DE genes \
and enriched pathways."Session continuity via --session-id ensures each turn builds on prior context. Results are stored in the .lobster_workspace/ directory and can be exported with /pipeline export.
What's Next?
Drug Resistance: Overcoming the BCR-ABL T315I Gatekeeper in CML
From target validation to resistance pharmacogenomics — how Lobster AI analyzes the canonical drug resistance story in chronic myeloid leukemia.
Genomics: From Variant QC to Clinical Prioritization
Population and clinical genomics across three complexity levels — variant QC, mitochondrial annotation, and a full clinical prioritization pipeline.